Sie befinden sich hier
Inhalt
What does “Sequencing Service” mean?
For researchers preparing their own sequencing libraries, the NGS core facility offers standalone next generation sequencing service.
What does “Full Service” mean?
For researchers which do not have the possibility to provide ready-to-sequence libraries, the NGS core facility offers a full service from nucleic acids, single cell or single nuclei to sequencing data. Researcher send the samples to NGS core facility where the samples will be processed in the wet lab and sequenced.
How do I submit the Sample Submission Sheet to provide detailed information about my samples?
Upon project confirmation, our NGS team will ask you to fill in our Sample Submission Sheet and send it back to us via email (ngs@medma.uni-heidelberg.de). Our team will update the details of the project on your behalf.
What happens after I submit a Sample Submission Sheet?
Our NGS team will review your request and guide you through the next steps.
What type of premade libraries can I submit?
You can submit any Illumina-compatible library for sequencing, such as libraries from genomic DNA for whole genome sequencing or RNA for transcriptome sequencing.
How do I prepare my libraries for sequencing?
You can use any Illumina-compatible library preparation kit.
What sequencing platforms and configurations are available?
We sequence on the Illumina instruments NovaSeq6000, NextSeq550) and MiSeq. The available sequencing modes are depending on the instrument and flow cell: 2x150 bp, 2x250 bp, 2x300 bp. Please contact us if your project requires a different platform or configuration.
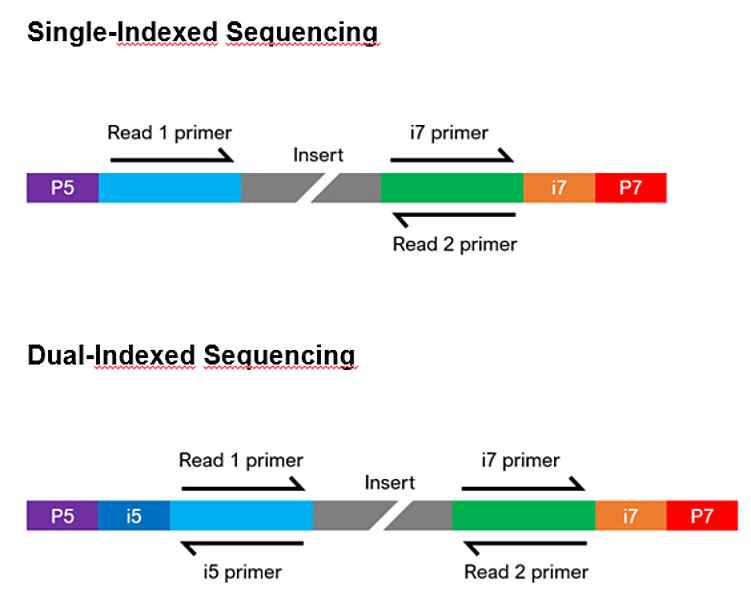
What does it mean to be a standard Illumina-compatible library?
Illumina-compatible libraries require 4 major components in their adapter sequences, found at the ends of each fragment, for paired-end sequencing:
- Flow cell binding sites. Known as P5 or P7, they facilitate clustering of the DNA strands on the flow cell.
- Sequencing primer binding sites. Labeled as Read 1 and Read 2, they are necessary to initiate sequencing at both ends of the fragment.
- Index sequences. Often called i5 and i7, they act as barcodes to allow multiplexing (pooling) of samples. Libraries can be either single index (having one index sequencing) or dual index (having two index sequences), and each library to be pooled requires at least one unique index.
- Index primer binding site(s). These are required to read the index sequences.
The composition of the adapter sequences depends on whether the library is single or dual indexed (see figure below). If any of the components are missing, heavily modified, or in the wrong configuration, sequencing may fail. If you have any questions regarding your library configuration, please contact us.
Can you spike PhiX into my sequencing run?
Yes, please instruct our team what percentage of PhiX spike-in to use, and we’ll accommodate the request free of charge. We typically recommend ~10% PhiX spike-in for projects with low diversity libraries.
Can you perform data analysis for my sequencing-only project?
Yes, please indicate this on the Sample Submission Sheet.
What are your starting material requirements for premade libraries?
We request that each library or library pool be ≥15 nM, ≥20 µL, in water or EB buffer. If you cannot meet these requirements, please contact us for further details.
How do I ship my premade libraries to you?
We request that you bring your libraries on ice or dry ice to our laboratory or ship your libraries overnight on dry ice to our laboratory (address details on contact us.
What QC is performed on the premade libraries?
We will run your libraries on the Agilent2100 Bioanalyzer or TapeStation to check the insert size and perform final quantification via Qubit prior to loading.
Can the NGS core facility de-multiplex my data?
We ask that you provide the index sequences in the Sample Submission Sheet prior to your sequencing run. Please contact us if our bioinformatics team shall perform the demultiplexing for you. Alternatively, if no index sequences are provided, we will deliver the raw data and you can perform the de-multiplexing.
Do you accept sequencing libraries with custom sequencing or index primers?
Yes, please contact us for further details.
How do I contact the NGS core facility for a technical consultation about my project?
Our scientists can help you optimize your project design and provide consultation. Please contact us.
How will my data be delivered?
Please contact us to find the ideal data delivery method for you.
How should I prepare and send my samples?
- Sample Type: Premade Sequencing Library
- Recommended Amount: >15 nM, >20 µL
- Buffer: Water, EB, or low TE (<0.1 mM EDTA)
- Shipment Method: Over night on dry ice or bring it to the facility on ice
Detailed information can be found in our Sample Submission and Sequencing Guidelines.
What happens if my samples do not meet your starting material requirements?
Please contact us.