Sie befinden sich hier
Inhalt
Our goal is to understand molecular disease mechanisms of heart failure to enable the development of novel therapeutic strategies, ultimately aiming to reduce mortality and morbidity in patients suffering from this common and debilitating disease.
Our interdisciplinary team of medical doctors, basic scientists, technicians and students employs modern cell and molecular biology methods as well as mouse models of human heart diseases, in order to address the following three related topics:
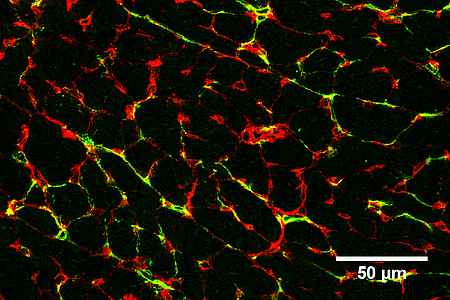
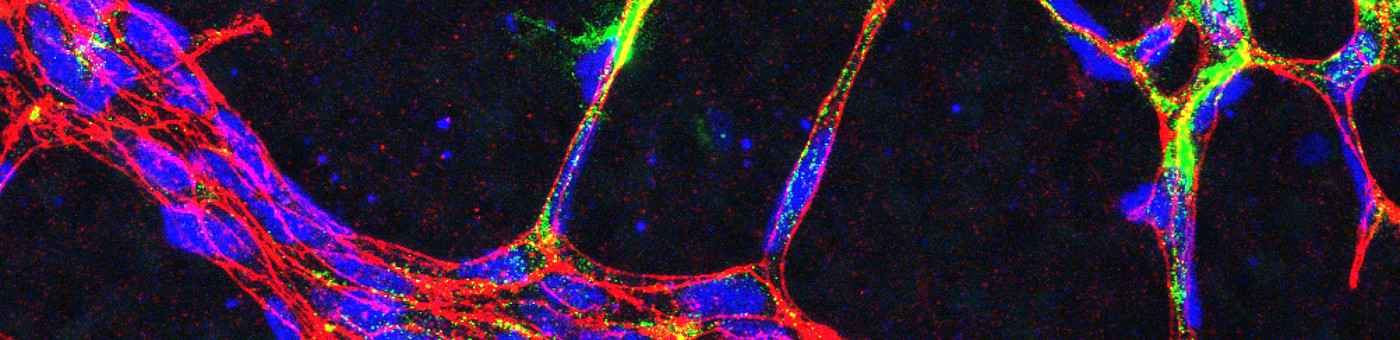
- We study the impact of myocardial intercellular interactions (for example between cardiomyocytes, endothelial cells and fibroblasts) during cardiac disease. We are especially interested in paracrine mechanisms and their upstream transcriptional regulators by which endothelial cell enable cardiac regeneration, promote cardiac adaptation and on the other hand drive the progression of heart failure.
- Epidemiological data imply a rising influence of primarily non-cardiac disease (such as cancer, diabetes or cachexia) on heart failure. In this regard, skeletal muscle wasting is associated with a dramatically worsened prognosis in heart failure. We systematically analyze the endocrine interaction between the heart and skeletal muscle in disease models and identified several mediators of this interaction that might serve as therapeutic target in the future.
- We probe cellular mechanisms of cardiomyocyte growth (“hypertrophy”), for example we address its regulation at the level of protein synthesis. Furthermore, we develop translational approaches to inhibit pathological cardiac hypertrophy via gene therapy or drugs.
Kontextspalte
Department of Cardiovascular Physiology
European Center for Angioscience (ECAS)
Medical Faculty Mannheim
Heidelberg University
Ludolf-Krehl-Straße 7 - 11
68167 Mannheim
Phone +49 621 383-71850
kphys@medma.uni-heidelberg.de
Head

Prof. Dr. Jörg Heineke