
Sie befinden sich hier
Inhalt
We provide data analysis services for:
Whole-exome sequencing (WES)

Exons make up about 1.5 % of the total genome in humans. Despite the low proportion of exons, these protein coding regions harbour up to 85% of disease-associated variations. Sequencing of exonic regions yields highly relevant genetic variants such as single nucleotide variations (SNVs), small insertions and deletions and structural aberrations that can help to determine the molecular landscape of human disorders.
To determine acquired variants that distinguish disease conditions from normal conditions, a test and a control sample (i. e. tumor and germline sample) are required for the analysis. Our data analysis pipeline comprises base calling (i.e. creation of fastq files), quality control and alignment of sequenced reads to the reference genome. Subsequent variant calling yields somatic mutations in VCF format which contains, among others, information about the genomic position, allele frequency and annotations of public databases regarding known disease-promoting effects of the identified variations. In addition, copy number analysis is performed to identify structural genomic variations like amplifications or deletions within exonic regions.
Contact: volker.ast@medma.uni-heidelberg.de
Single-cell RNA sequencing (SC RNA-seq)
Single-cell RNA sequencing (scRNA-seq) is a powerful technique to uncover the heterogeneous composition of a given tissue on the cellular level and to reveal the contribution of diverse cell populations to the tissue’s complex expression pattern. ScRNA-seq is also applied to dissect the trajectories of different cell lineages in various states of differentiation or development.
State-of-the-art analysis of scRNA-seq comprises doublet detection, normalization, dimension reduction by principal component analysis (PCA) and clustering using different methods (i. e. tSNE or UMAP). Cell types are identified by integrating clustering and gene expression data with subsequent evaluation of the expression profiles of known marker genes.
In addition, immune cell profiling can be performed using the 10x GenomicsTM V(D)J enrichment technology to uncover a sample’s lymphocyte repertoire. In detail, T cell diversity is determined by identifying full-length α and β chain sequences of T-cell receptors whereas B cell diversity is revealed by identifying full-length light and heavy chain antibody sequences.
.")
.")
Contact: volker.ast@medma.uni-heidelberg.de
RNA sequencing (RNA-seq)
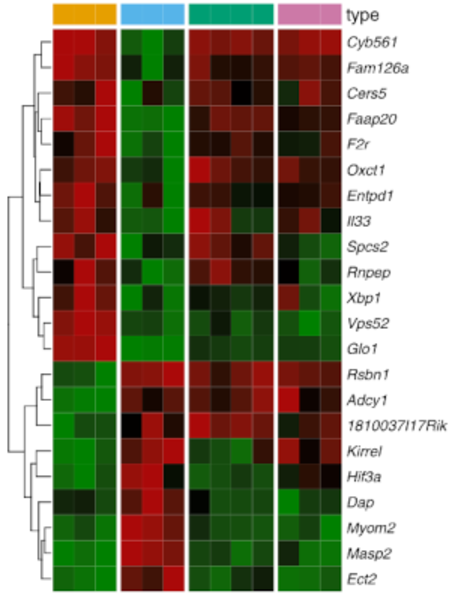
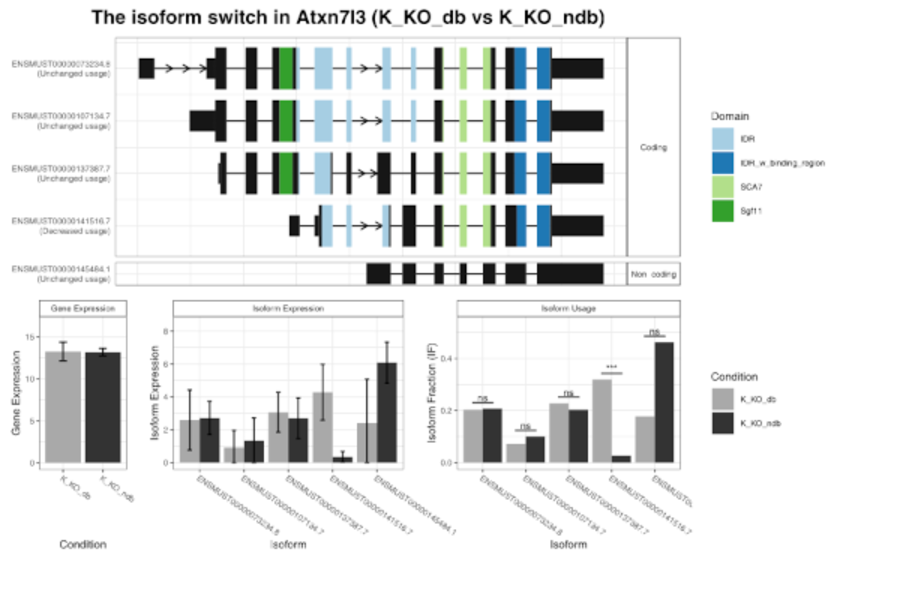
MRNA-Seq for gene expression analysis, detection of splicing events and fusion genes. Gain one of the most comprehensive views of all expressed transcripts within your sample with our RNA-Seq services. Since the entire transcriptome is covered with short Illumina reads, RNA-Seq is the preferred method to analyse novel transcripts, novel isoforms, alternative splice sites, rare transcripts or fusion genes.
Chromatin ImmunoPrecipitation DNA sequencing (ChIP-seq)
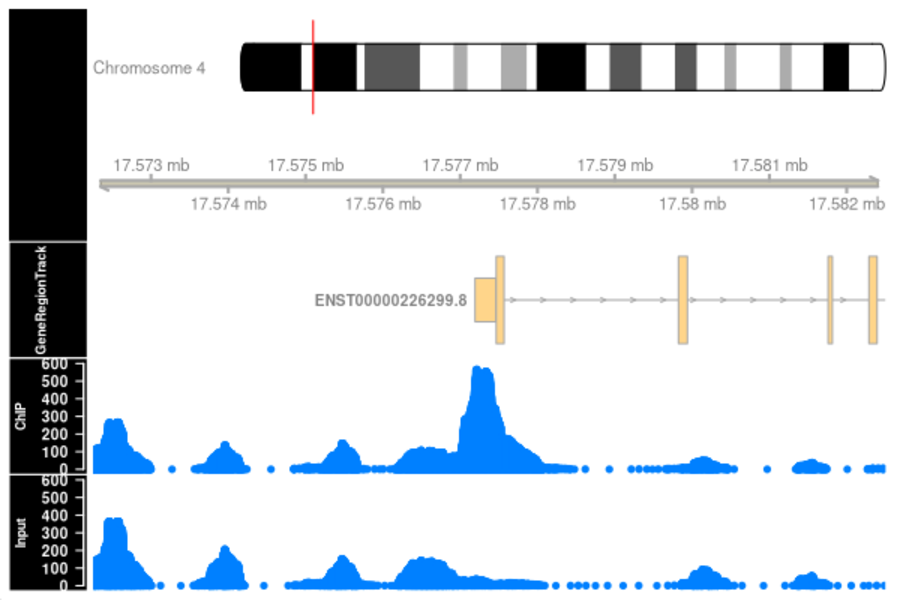
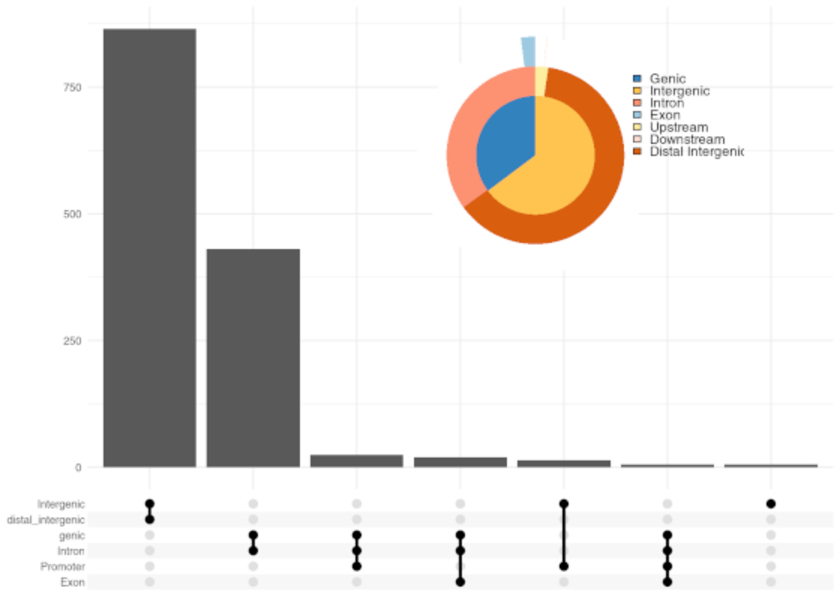
ChIP-Seq (Chromatin ImmunoPrecipitation DNA-Sequencing) is a biochemical method for the determination of protein-DNA interactions. ChIP-Seq is a combination of chromatin immunoprecipitation and high-throughput DNA sequencing. DNA-protein interactions occur in binding sequences for transcription factors on promoters, enhancers, repressors, silencers, isolators and in binding sequences for DNA replication.
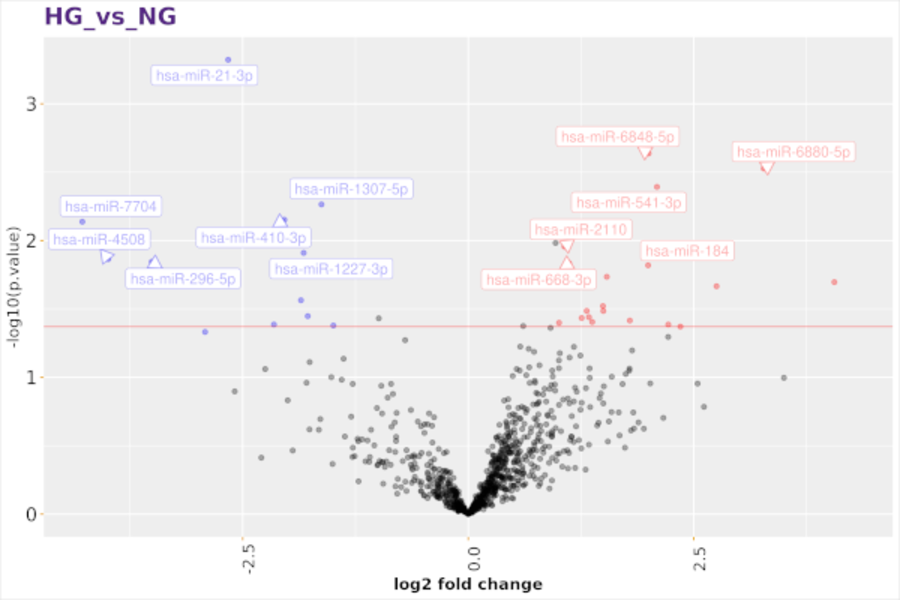
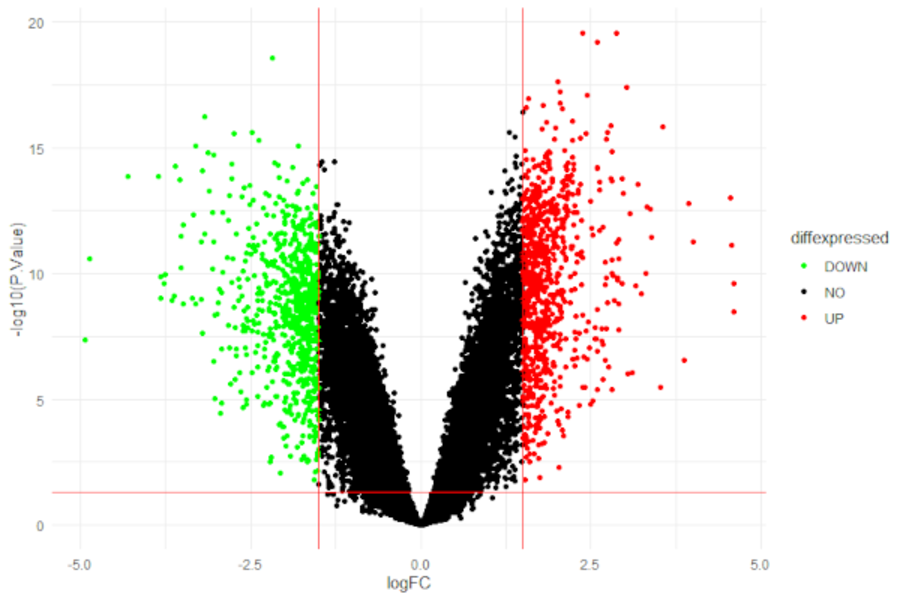
Micro-RNA sequencing (miRNA-seq)
Discover new small RNAs and analyse the expression of known small RNAs. Small RNAs constitute a family of RNA species that plays a central role in regulating gene expression in many organisms. With the enormous depth reached by next generation sequencing technologies, you can get a comprehensive view of the small RNAs in your samples. Sequencing small RNA is done by preparing a short insert cDNA library for each sample.
Typical projects are:
- differential expression analysis of small RNAs
- identification of novel small RNAs
- identification of target molecules of novel miRNAs
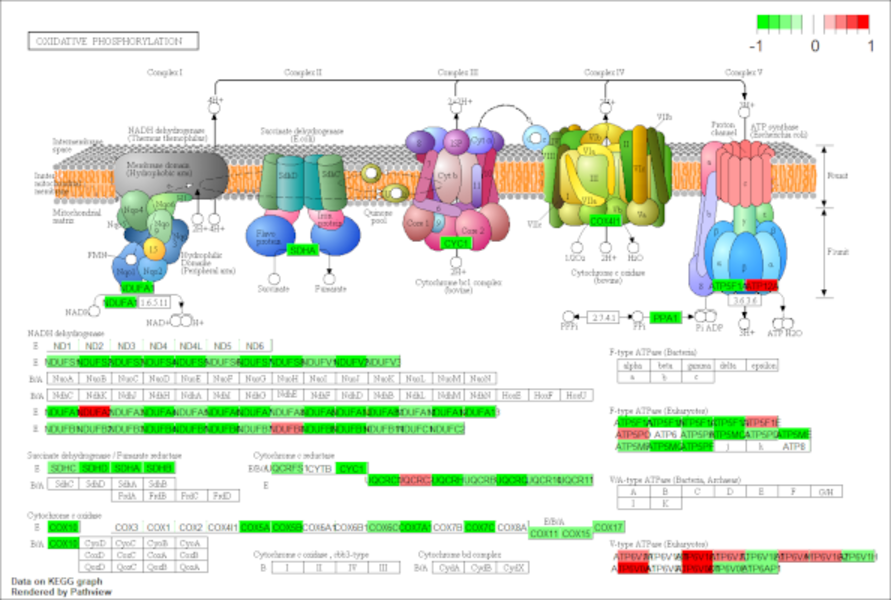
Affymetrix microarray analysis
The Affymetrix core facility is available to perform gene expression analysis. We not only perform the experiments, but also give advice and solve all the doubts that may arise in the process, from the setting up of the experimental design, to the writing of manuscripts. We are in constant and close contact with the Affymetrix field specialists in order to know the latest developments in the available technology. In this way, and thanks to our experience, we can give advice to which are the chips that better adapt according to the needs of each particular experiment.
Further tasks we can help you with are:
- Assist your doubt about experimental design.
- Carry out the quality control of the samples.
- Perform the protocols of hybridization of microarrays.
- Data analysis (standardization, statistical analysis, pathway analysis and GSEA, among others) and visualization.
- Text, manuscripts and scientific papers writing.
- Data base submission (e. g. GEO)
If you have any other requests, contact us and let us know. We will be looking forward to helping you out.
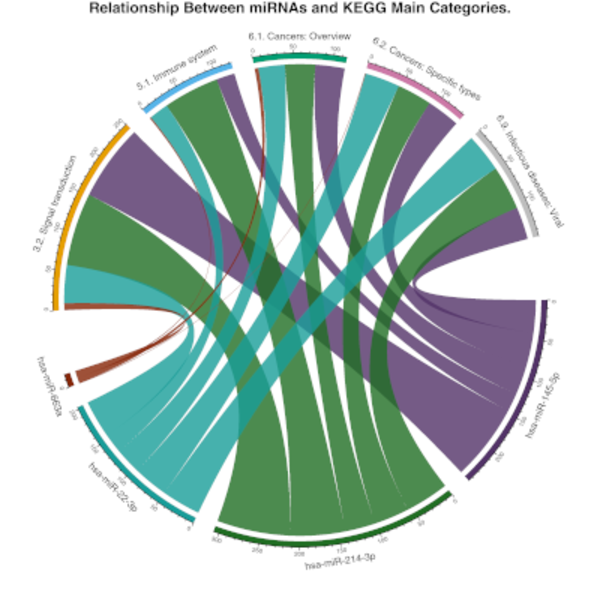
Micro-RNA target gene prediction
miRWalk is an improved version of the previous database (i. e. miRWalk). The new version of miRWalk stores predicted data obtained with a maschine learning algorithm including experimentally verified miRNA-target interactions.